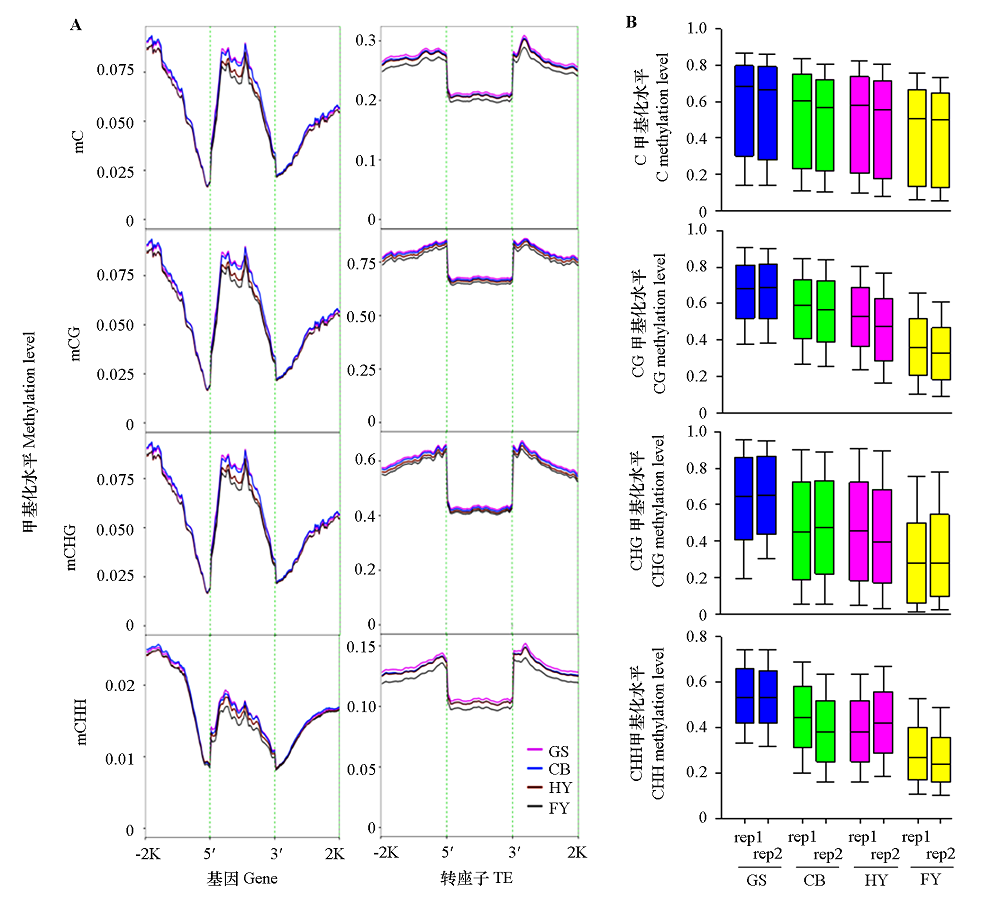
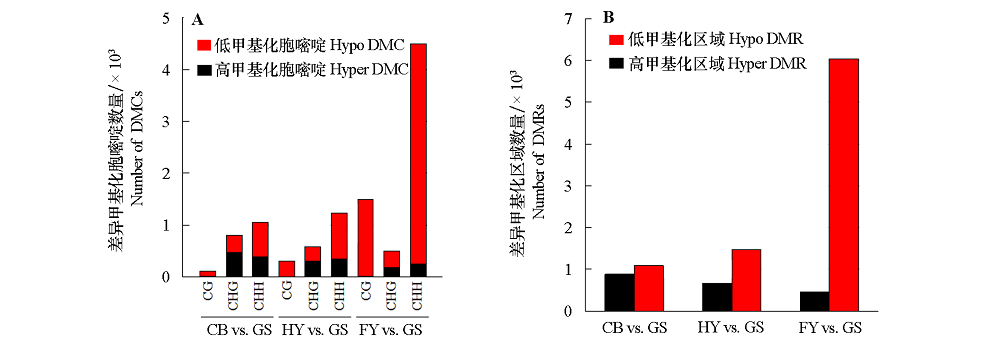
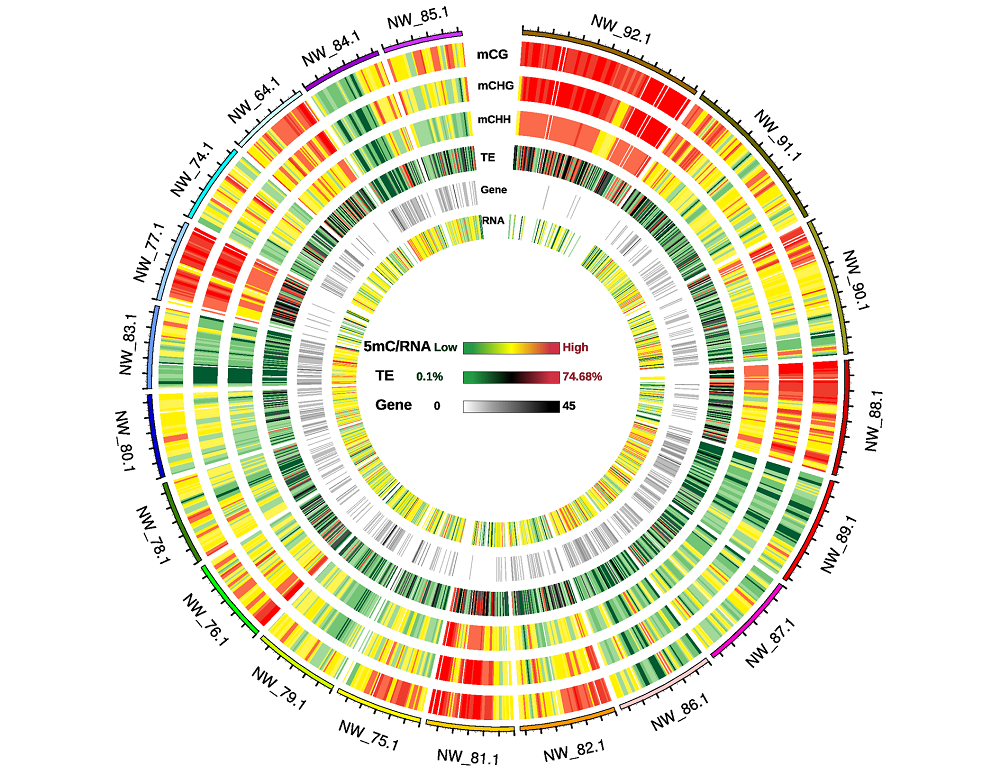
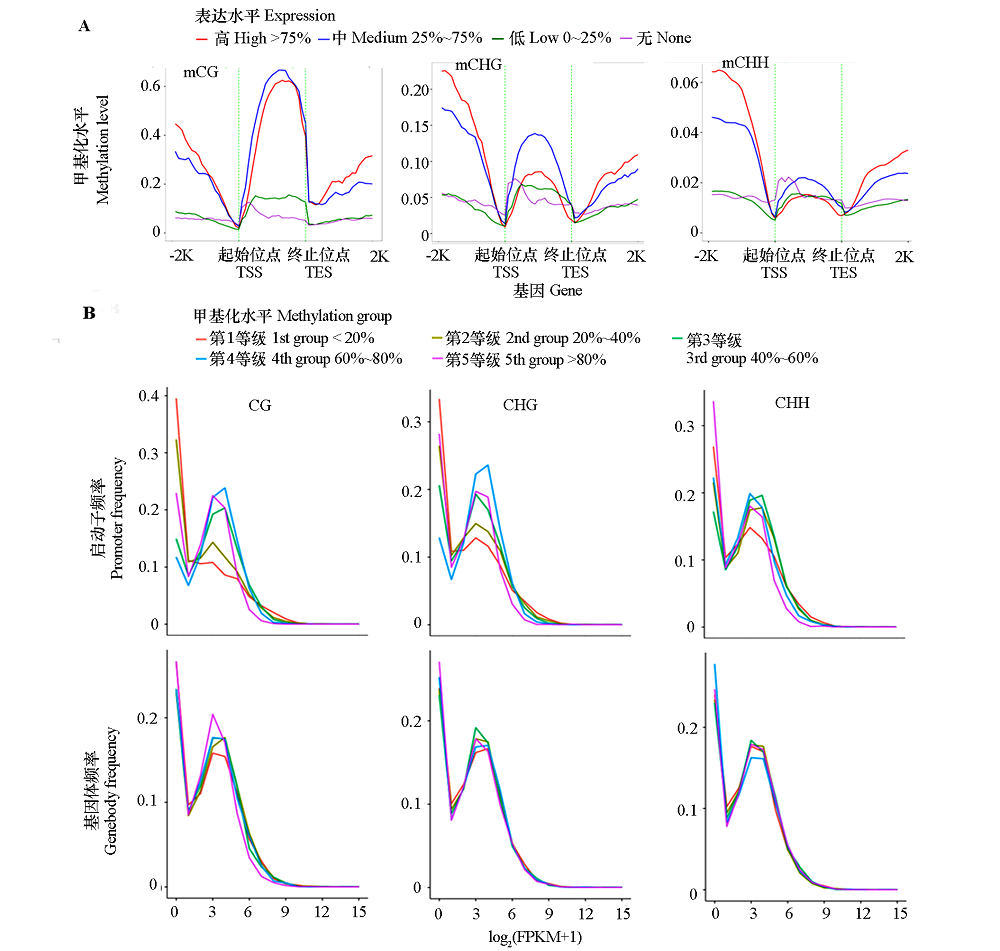
| [1] |
Ahmad A, Zhang Y, Cao X F. 2010. Decoding the epigenetic language of plant development. Molecular Plant, 3(4):719-728.
doi: 10.1093/mp/ssq026
pmid: 20663898
|
| [2] |
Bolger A M, Lohse M, Usadel B. 2014. Trimmomatic:a flexible trimmer for Illumina sequence data. Bioinformatics, 30(15):2114-2120.
doi: 10.1093/bioinformatics/btu170
URL
|
| [3] |
Bonasio R, Tu S, Reinberg D. 2010. Molecular signals of epigenetic states. Science, 330(6004):612-616.
doi: 10.1126/science.1191078
URL
|
| [4] |
Chen Duanfen, Peng Zhenhua, Gao Zhimin. 2008. Cloning and expression analysis of PDS gene in Narcissus tazetta var. chinensis. Molecular Plant Breeding, 6(3):574-578. (in Chinese)
|
|
陈段芬, 彭镇华, 高志民. 2008. 中国水仙八氢番茄红素脱氢酶基因(PDS)的克隆及表达分析. 分子植物育种, 6(3):574-578.
|
| [5] |
Chen Yong-ping, Gao Feng, Shen Yan-hong, Zhao Wan-wan, Chen Gui-xin, Wang Ping. 2019. The analysis of ERFs related to fruit ripening in papaya. Acta Horticulturae Sinica, 46(2):252-264. (in Chinese)
|
|
陈永萍, 高峰, 申艳红, 赵弯弯, 陈桂信, 王平. 2019. 番木瓜ERF家族与果实成熟相关成员的分析. 园艺学报, 46(2):252-264.
|
| [6] |
Cheng J, Niu Q, Zhang B, Chen K, Yang R, Zhu J K, Zhang Y, Lang Z. 2018. Downregulation of RdDM during strawberry fruit ripening. Genome Biology, 19(1):212.
doi: 10.1186/s13059-018-1587-x
URL
|
| [7] |
Ding X, Zhu X, Ye L, Xiao S, Wu Z, Chen W, Li X. 2019. The interaction of CpEBF1 with CpMADSs is involved in cell wall degradation during papaya fruit ripening. Horticulture Research, 6(1):13.
doi: 10.1038/s41438-018-0095-1
URL
|
| [8] |
Du Xiaoyun, Song Laiqing, Zhao Lingling, Liu Meiying, Tang Yan, Sun Yanxia, Jiang Zhongwu, Shu Huairui. 2019. DNA methylation in Red Fuji apple bud sports lines. Acta Horticulturae Sinica, 46(1):107-120. (in Chinese)
|
|
杜晓云, 宋来庆, 赵玲玲, 刘美英, 唐岩, 孙燕霞, 姜中武, 束怀瑞. 2019. 红富士苹果芽变系DNA甲基化研究. 园艺学报, 46(1):107-120.
|
| [9] |
Fabi J P, Do Prado S B R. 2019. Fast and furious:ethylene-triggered changes in the metabolism of papaya fruit during ripening. Frontiers in Plant Science, 10:535.
doi: 10.3389/fpls.2019.00535
URL
|
| [10] |
Fan Zhong-qi, Kuang Jian-fei, Lu Wang-jin, Chen Jian-ye. 2015. Advances in research of the mechanism of transcription factors involving in regulating fruit ripening and senescence. Acta Horticulturae Sinica, 42(9):1649-1663. (in Chinese)
|
|
范中奇, 邝健飞, 陆旺金, 陈建业. 2015. 转录因子调控果实成熟和衰老机制研究进展. 园艺学报, 42(9):1649-1663.
|
| [11] |
Finnegan E J, Kovac K A. 2000. Plant DNA methyltransferases. Plant Molecular Biology, 43:189-201.
pmid: 10999404
|
| [12] |
Grafi G. 2011. Epigenetics in plant development and response to stress. Biochimica et Biophysica Acta, 1809:351-352.
|
| [13] |
He X J, Chen T, Zhu J K. 2011. Regulation and function of DNA methylation in plants and animals. Cell Research, 21:442-465.
doi: 10.1038/cr.2011.23
URL
|
| [14] |
Huang H, Liu R, Niu Q, Tang K, Zhang B, Zhang H, Chen K, Zhu J K, Lang Z. 2019. Global increase in DNA methylation during orange fruit development and ripening. Proceedings of the National Academy of Sciences of the United States of America, 116(4):1430-1436.
|
| [15] |
Huang Wen-jun, Zhong Cai-hong. 2017. Research advances in the postharvest physiology of kiwifruit. Plant Science Journal, 35(4):622-630. (in Chinese)
|
|
黄文俊, 钟彩虹. 2017. 猕猴桃果实采后生理研究进展. 植物科学学报, 35(4):622-630.
|
| [16] |
Krueger F, Andrews S R. 2011. Bismark:a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics, 27(11):1571-1572.
doi: 10.1093/bioinformatics/btr167
pmid: 21493656
|
| [17] |
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones S J, Marra M A. 2009. Circos:an information aesthetic for comparative genomics. Genome Research, 19:1639-1645.
doi: 10.1101/gr.092759.109
pmid: 19541911
|
| [18] |
Lang Z, Wang Y, Tang K, Tang D, Datsenka T, Cheng J, Zhang Y, Handa A K, Zhu J K. 2017. Critical roles of DNA demethylation in the activation of ripeninginduced genes and inhibition of ripening-repressed genes in tomato fruit. Proceedings of the National Academy of Sciences of the United States of America, 114:E4511-E4519.
|
| [19] |
Law J A, Jacobsen S E. 2010. Establishing,maintaining and modifying DNA methylation patterns in plants and animals. Nature Reviews Genetics, 11:204-220.
doi: 10.1038/nrg2719
URL
|
| [20] |
Li X, Zhu J, Hu F, Ge S, Ye M, Xiang H, Zhang G, Zheng X, Zhang H, Zhang S, Li Q, Luo R, Yu C, Yu J, Sun J, Zou X, Cao X, Xie X, Wang J, Wang W. 2012. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genomics, 13:300.
doi: 10.1186/1471-2164-13-300
URL
|
| [21] |
Lister R, O’Malley R C, Tonti-Filippini J, Gregory B D, Berry C C, Millar A H, Ecker J R. 2008. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell, 133:523-536.
doi: 10.1016/j.cell.2008.03.029
pmid: 18423832
|
| [22] |
Liu R, How-Kit A, Stammitti L, Teyssier E, Rolin D, Mortain-Bertrand A, Halle S, Liu M, Kong J, Wu C, Degraeve-Guibault C, Chapman N H, Maucourt M, Hodgman T C, Tost J, Bouzayen M, Hong Y, Seymour G B, Giovannoni J J, Gallusci P. 2015. A DEMETER-like DNA demethylase governs tomato fruit ripening. Proceedings of the National Academy of Sciences of the United States of America, 112:10804-10809.
|
| [23] |
Liu Xin, Chen Yunzhu, Kim Pyol, Kim Min-Jun, Song Hyondok, Li Yuhua, Wang Yu. 2020. Progress on molecular mechanism and regulation of tomato fruit color formation. Acta Horticulturae Sinica, 47(9):1689-1704. (in Chinese)
|
|
刘昕, 陈韵竹, Kim Pyol, Kim Min-Jun, Song Hyondok, 李玉花, 王宇. 2020. 番茄果实颜色形成的分子机制及调控研究进展. 园艺学报, 47(9):1689-1704.
|
| [24] |
Lu X, Wang X, Chen X, Shu N, Wang J, Wang D, Wang S, Fan W, Guo L, Guo X, Ye W. 2017. Single-base resolution methylomes of upland cotton(Cossypium hirsutum L.)reveal epigenome modification in response to drought stress. BMC Genomics, 18(1):297.
doi: 10.1186/s12864-017-3681-y
URL
|
| [25] |
Manning K, Tör M, Poole M, Hong Y, Thompson A J, King G J, Giovannoni J J, Seymour G B. 2006. A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nature Genetics, 38(8):948.
doi: 10.1038/ng1841
URL
|
| [26] |
Marty I, Bureau S, Sarkissian G, Gouble B, Audergon J M, Albagnac G. 2005. Ethylene regulation of carotenoid accumulation and carotenogenic gene expression in colour-contrasted apricot varieties(Prunus armeniaca). Journal of Experimental Botany, 56(417):1877-1886.
pmid: 15911563
|
| [27] |
Muñoz-Bertomeu J, Miedes E, Lorences E P. 2013. Expression of xyloglucan endotransglucosylase/hydrolase(XTH)genes and XET activity in ethylene treated apple and tomato. Journal of Plant Physiology, 170(13):1194-1201.
doi: 10.1016/j.jplph.2013.03.015
pmid: 23628624
|
| [28] |
Narsaiah K, Wilson R A, Gokul K, Mandge H M, Jha S N, Bhadwal S, Anurag R K, Malik RK, Vij S. 2015. Effect of bacteriocin-incorporated alginate coating on shelf-life of minimally processed papaya(Carica papaya L.). Postharvest Biology and Technology, 100:212-218.
doi: 10.1016/j.postharvbio.2014.10.003
URL
|
| [29] |
Niederhuth C E, Bewick A J, Ji L, Alabady M S, Kim K D, Li Q, Rohr N A, Rambani A, Burke J M, Udall J A, Egesi C, Schmutz J, Grimwood J, Jackson S A, Springer N M, Schmitz R J. 2016. Widespread natural variation of DNA methylation within angiosperms. Genome Biology, 17:194.
pmid: 27671052
|
| [30] |
Pesaresi P, Mizzotti C, Colombo M, Masiero S. 2014. Genetic regulation and structural changes during tomato fruit development and ripening. Frontiers in Plant Science, 23(5):124.
|
| [31] |
Pfaffl M W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research, 29(9):e45.
doi: 10.1093/nar/29.9.e45
pmid: 11328886
|
| [32] |
Qi W, Wang H, Zhou Z, Yang P, Wu W, Li Z, Li X. 2020. Ethylene emission as a potential indicator of Fuji apple flavor quality evaluation under low temperature. Horticultural Plant Journal, 6(4):231-239.
doi: 10.1016/j.hpj.2020.03.007
URL
|
| [33] |
Seymour DK, Koenig D, Hagmann J, Becker C, Weigel D. 2014. Evolution of DNA methylation patterns in the Brassicaceae is driven by differences in genome organization. PLoS Genetics, 10:e1004785.
|
| [34] |
Shen Y H, Lu B G, Feng L, Yang F Y, Geng J J, Ming R, Chen X J. 2017. Isolation of ripening-related genes from ethylene/1-MCP treated papaya through RNA-seq. BMC Genomics, 18(1):671.
doi: 10.1186/s12864-017-4072-0
URL
|
| [35] |
Tang K, Lang Z, Zhang H, Zhu J K. 2016. The DNA demethylase ROS1 targets genomic regions with distinct chromatin modifications. Nature Plants, 2:16169.
doi: 10.1038/nplants.2016.169
pmid: 27797352
|
| [36] |
Tian Shi-ping. 2013. Molecular mechanisms of fruit ripening and senescence. Chinese Bulletin of Botany, 48(5):481-488. (in Chinese)
doi: 10.3724/SP.J.1259.2013.00481
URL
|
|
田世平. 2013. 果实成熟和衰老的分子调控机制. 植物学报, 48(5):481-488.
|
| [37] |
Wang Zhong-feng. 2009. Research advancement in relation of enzymes for cell wall metabolism with fruit softening. Chinese Agricultural Science Bulletin, 25(18):126-130. (in Chinese)
|
|
王中凤. 2009. 细胞壁分解酶与果实软化的关系研究进展. 中国农学通报, 25(18):126-130.
|
| [38] |
Xu J, Wang X, Cao H, Xu H, Xu Q, Deng X. 2017. Dynamic changes in methylome and transcriptome patterns in response to methyltransferase inhibitor 5-azacytidine treatment in citrus. DNA Research, 24(5):509-522.
doi: 10.1093/dnares/dsx021
URL
|
| [39] |
Xu J, Zhou S, Gong X, Song Y, van Nocker S, Ma F, Guan Q. 2018. Single-base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnology Journal, 16:672-687.
doi: 10.1111/pbi.2018.16.issue-2
URL
|
| [40] |
Xu Sheng, Zhang Xin, Liu Decai, Gu Tingting. 2020. Identification and functional analysis of N6-adenine methylation(6mA)methyltransferase genes in Fragaria vesca. Acta Horticulturae Sinica, 47(11):2194-2206. (in Chinese)
|
|
徐昇, 张鑫, 刘德才, 顾婷婷. 2020. 森林草莓6mA甲基转移酶基因鉴定及功能分析. 园艺学报, 47(11):2194-2206.
|
| [41] |
Zhang M, Zhang X, Guo L, Qi T, Liu G, Feng J, Shahzad K, Zhang B, Li X, Wang H, Tang H, Qiao X, Wu J, Xing C. 2020. Single-base resolution methylome of cotton cytoplasmic male sterility system reveals epigenomic changes in response to high-temperature stress during anther development. Journal of Experimental Botany, 71:951-969.
doi: 10.1093/jxb/erz470
pmid: 31639825
|
| [42] |
Zhang X. 2008. The epigenetic landscape of plants. Science, 320(5875):489-492.
doi: 10.1126/science.1153996
URL
|
| [43] |
Zhong S, Fei Z, Chen Y R, Zheng Y, Huang M, Vrebalov J, McQuinn R, Gapper N, Liu B, Xiang J, Shao Y, Giovannoni J J. 2013. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nature Biotechnology, 31(2):154-159.
doi: 10.1038/nbt.2462
URL
|
| [44] |
Zhou C, Yang M, Luo X, Kuang R, Yang H, Yao J, Huang B, Wei Y. 2021. Transcriptomic analysis of papaya(Carica papaya L.)shoot explants obtained by leaf- and stem-inoculation methods for adventitious roots induction. Scientia Horticulturae, 276:109762.
|
| [45] |
Zhu J K. 2009. Active DNA demethylation mediated by DNA glycosylases. Annual Review of Genetics, 43:143-66.
doi: 10.1146/genet.2009.43.issue-1
URL
|