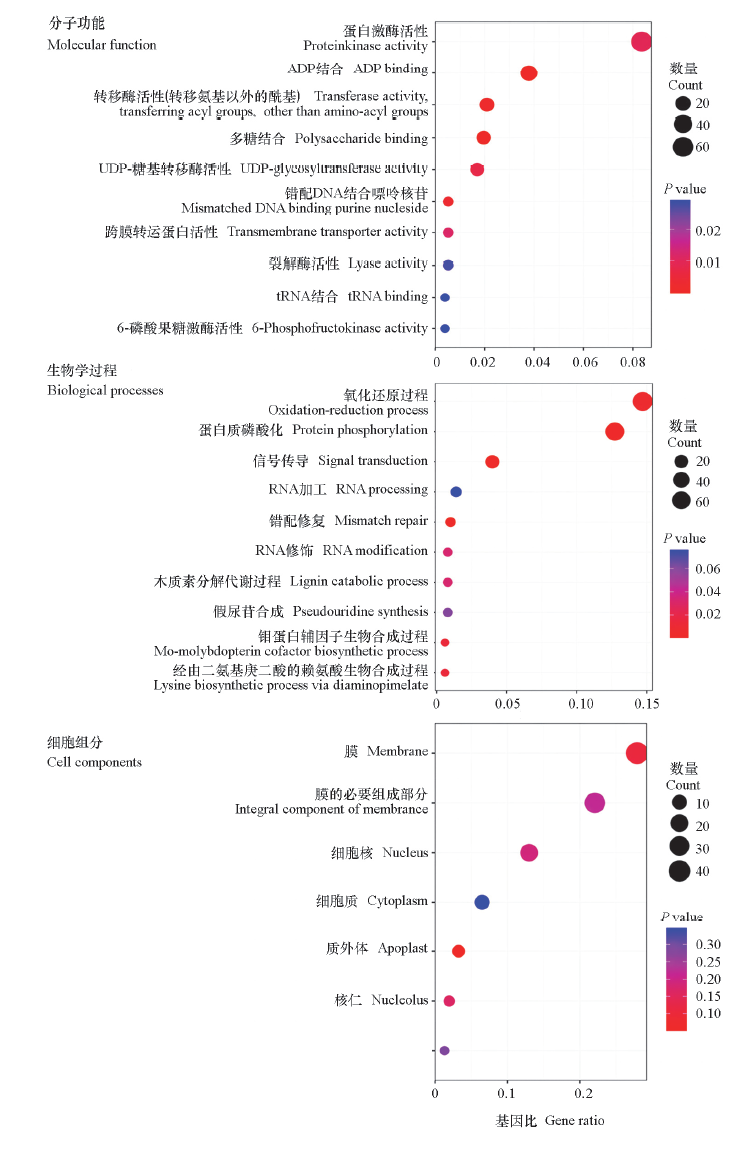
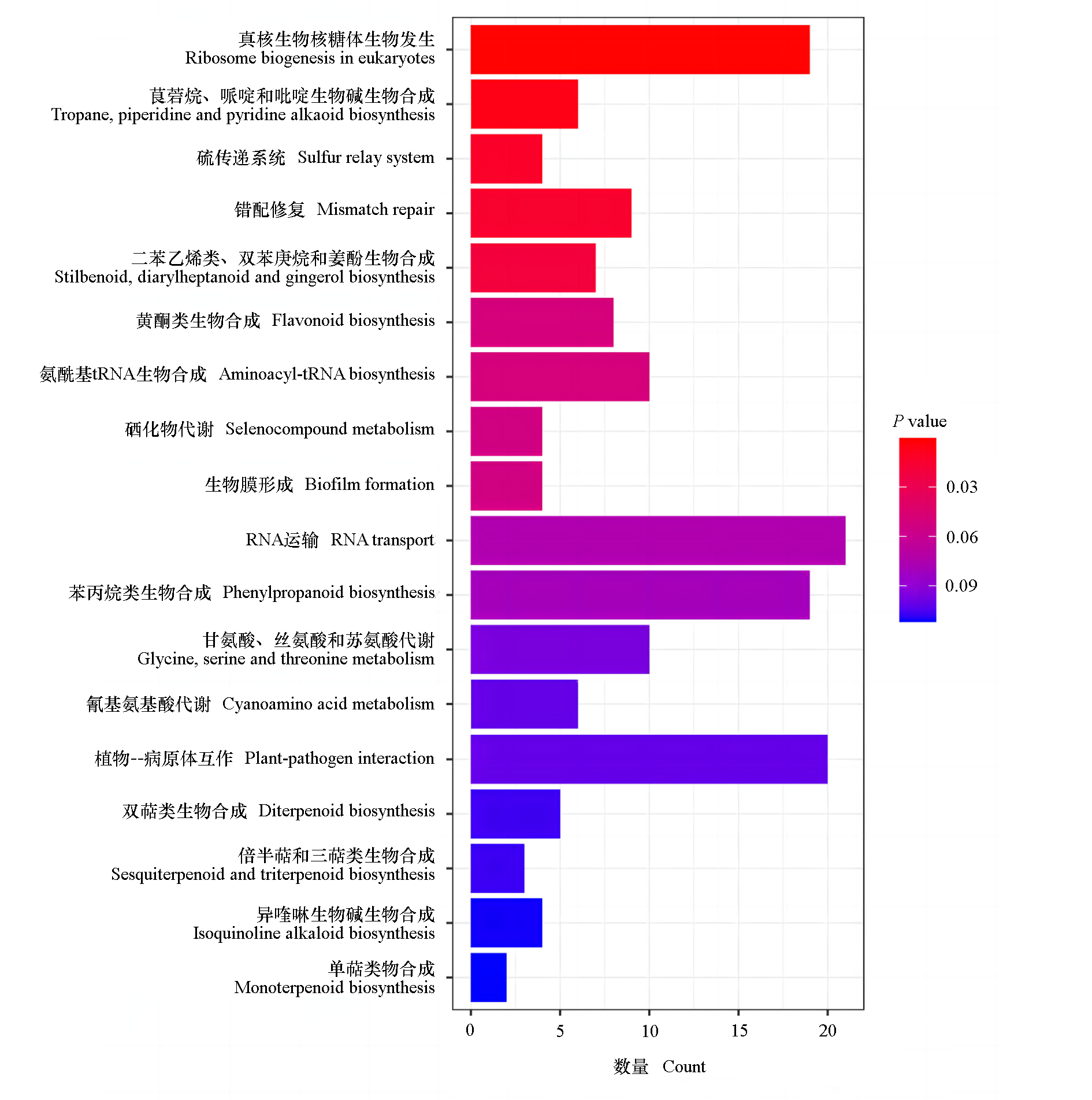
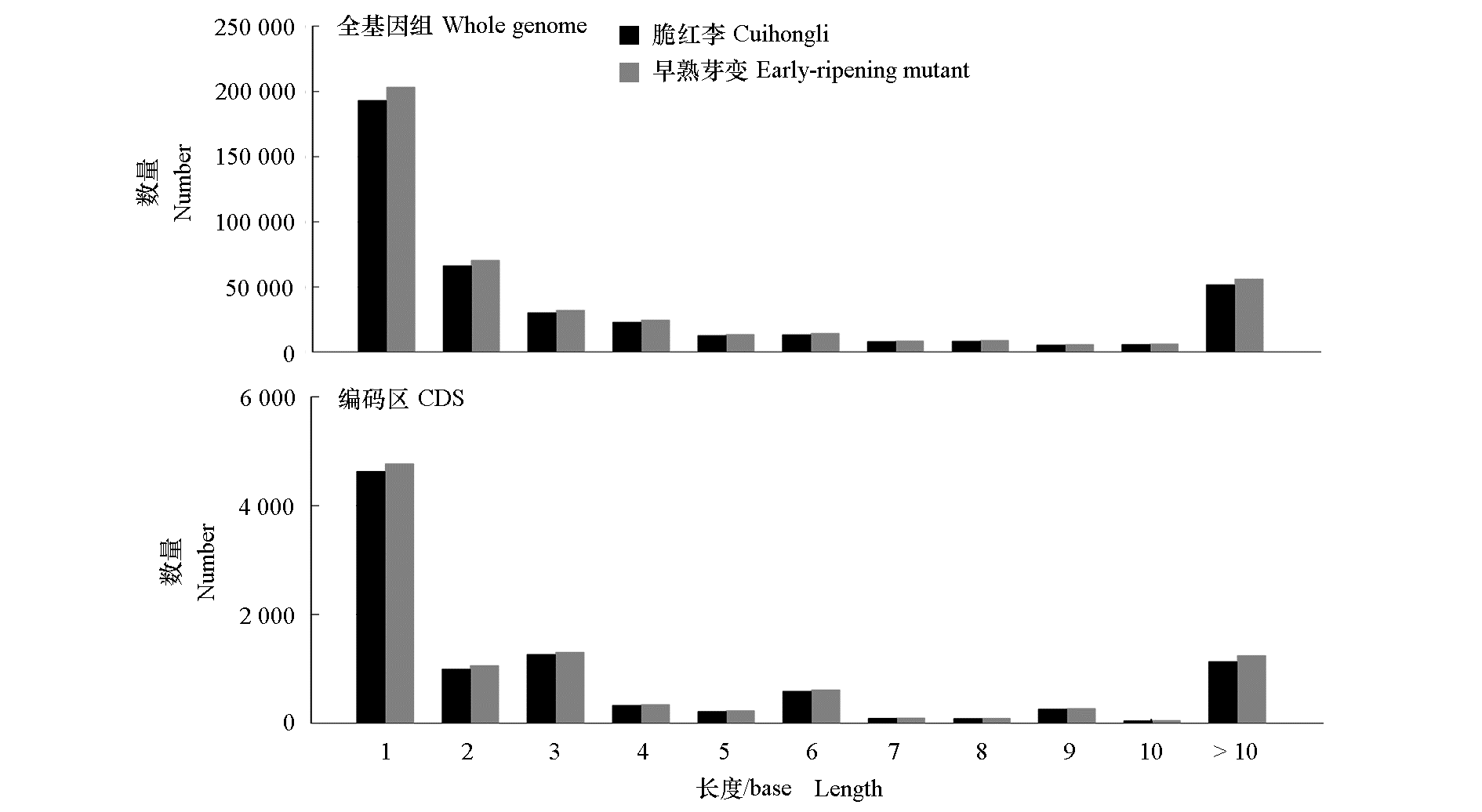| [1] |
Ashburner M, Ball C A, Blake J A, Botstein D, Butler H, Cherry J M, Davis A P, Dolinski K, Dwight S S, Eppig J T, Harris M A, Hill D P, Issel-Tarver L, Kasarskis A, Lewis S, Matese J C, Richardson J E, Ringwald M, Rubin G M, Sherlock G. 2000. Gene ontology:tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics, 25 (1):25-29.
doi: 10.1038/75556
pmid: 10802651
|
| [2] |
Aslam M M. 2021. Molecular mechanisms of sugar metabolic and ABA signal transduction key genes in regulating peach fruit ripening[Ph. D. Dissertation]. Beijing: Chinese Academy of Agricultural Sciences.
|
| [3] |
Chang Ailing. 2017. Development of InDel and SV markers for Citrus suavissima‘Wuzi Qugan’[M. D. Dissertation]. Hangzhou: Zhejiang A & F University. (in Chinese)
|
|
常爱玲. 2017. ‘无子瓯柑’InDel和SV标记开发[硕士论文]. 杭州: 浙江农林大学.
|
| [4] |
Cui Lulu. 2021. Analysis of genetic characteristics of‘Licheng’based on molecular marker and genome resequencing[M. D. Dissertation]. Chongqing: Southwest University. (in Chinese)
|
|
崔璐璐. 2021. 基于分子标记及基因组重测序的‘梨橙’遗传特性分析[硕士论文]. 重庆: 西南大学.
|
| [5] |
Dong Yiling, Xiao Xuteng, Zhang Min, Shen Leyi, Chen Tianchi, Jia Yonghong, Wu Yueyan. 2022. The effect of ringing on the expression of VvNCED gene and fruit ripening in grapes. Journal of Nuclear Agricultural Sciences, 36 (7):1339-1349. (in Chinese)
doi: 10.11869/j.issn.100-8551.2022.07.1339
|
|
董昳伶, 肖旭腾, 张敏, 沈乐意, 陈天池, 贾永红, 吴月燕. 2022. 环割对葡萄VvNCED基因的表达和果实成熟的影响. 核农学报, 36 (7):1339-1349.
doi: 10.11869/j.issn.100-8551.2022.07.1339
|
| [6] |
Ge Yu, Xu Zining, Ma Weihong, Liu Yuanzheng, Wang Butian, Liu Yi. 2022. Genome-wide variation and phylogenetic analysis of different ecotypes of avocado based on whole genome re-sequencing. Chinese Journal of Tropical Agriculture, 42 (11):50-57. (in Chinese)
|
|
葛宇, 徐梓宁, 马蔚红, 刘远征, 王步天, 刘毅. 2022. 基于重测序的不同生态型油梨的全基因组变异及进化分析. 热带农业科学, 42 (11):50-57.
|
| [7] |
Hunt R C, Simhadri V L, Iandoli M, Sauna Z E, Kimchi-Sarfaty C. 2014. Exposing synonymous mutations. Trends in Genetics, 30 (7):308-321.
doi: 10.1016/j.tig.2014.04.006
pmid: 24954581
|
| [8] |
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. 2004. The KEGG resource for deciphering the genome. Nucleic Acids Research,32 (Database issue):D277-D280.
|
| [9] |
Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics, 25 (14):1754-1760.
|
| [10] |
Li P M, Zhang Y Z, Einhorn T C, Cheng L L. 2014. Comparison of phenolic metabolism and primary metabolism between green‘Anjou’pear and its bud mutation,red‘Anjou’. Physiologia Plantarum, 150 (3):339-354.
|
| [11] |
Li W F, Mao J, Yang S J, Guo Z G, Ma Z H, Dawuda M M, Zuo C W, Chu M Y, Chen B H. 2018. Anthocyanin accumulation correlates with hormones in the fruit skin of‘Red Delicious’and its four generation bud sport mutants. BMC Plant Biology,18:363.
|
| [12] |
Li Xingwu, Zhang Lili. 2018. Effects of different sterilization methods on nutritional ingredients and sensory quality of low-sugar Prunus salicina Jam. Food Research and Development, 39 (11):70-75. (in Chinese)
|
|
李兴武, 章黎黎. 2018. 不同杀菌方式对‘脆红李’低糖果酱营养成分和感官品质的影响. 食品研究与开发, 39 (11):70-75.
|
| [13] |
Li Xingwu, Zhang Lili. 2019. Optimization of hot air combined with ethanol fumigation on preservation of Prunus salicina cv.‘Cuihongli’. Storage and Process, 19 (6):34-39. (in Chinese)
|
|
李兴武, 章黎黎. 2019. 热空气结合乙醇熏蒸保鲜‘脆红李’的工艺优化. 保鲜与加工, 19 (6):34-39.
|
| [14] |
Lin Yao. 2019. Genome resequencing dataset based SSR locus analysis and its application in identification and evaluation of peach(Prunus persica)wild resources[M. D. Dissertation]. Nanjing: Nanjing Agricultural University. (in Chinese)
|
|
林瑶. 2019. 基于重测序的桃SSR位点分析及其在野生资源鉴定评价中的应用[硕士论文]. 南京: 南京农业大学.
|
| [15] |
Li Feifei. 2020. Study on the main characteristics and formation mechanism of fruit quality of‘Zaomi’Ponkan[Ph. D. Dissertation]. Changsha: Hunan Agricultural University. (in Chinese)
|
|
李菲菲. 2020. ‘早蜜’椪柑主要品质特征及其形成机理的研究[博士论文]. 长沙: 湖南农业大学.
|
| [16] |
Liu Q, Xu J, Liu Y Z, Zhao X L, Deng X X, Guo L L, Gu J Q. 2007. A novel bud mutation that confers abnormal patterns of lycopene accumulation in sweet orange fruit(Citrus sinensis L. Osbeck). Journal of Experimental Botany, 58 (15-16):4161-4171.
|
| [17] |
Mckenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo M A. 2010. The genome analysis toolkit:a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Research, 20 (9):1297-1303.
|
| [18] |
Nwafor C C, Gribaudo I, Schneider A, Wehrens R, Grando M S, Costantini L. 2014. Transcriptome analysis during berry development provides insights into co-regulated and altered gene expression between a seeded wine grape variety and its seedless somatic variant. BMC Genomics,Doi: 10.1186/1471-2164-15-1030.
|
| [19] |
Noda T, Daiou K, Mihara T, Nagano Y. 2020. Development of InDel markers for the selection of Satsuma mandarin(Citrus unshiu Marc.) hybrids that can be used for low-cost genotyping with agarose gels. Euphytica, 216 (7):115.
|
| [20] |
Petit J R, Hampe A. 2006. Some evolutionary consequences of being a tree. Annual Review of Ecology,Evolution,and Systematics,37:187-214.
|
| [21] |
Qiao Zhenglin, Hu Huizhen, Yan Bo, Chen Longqing. 2021. Advances of researches on biosynthesis and regulation of floral volatile benzenoids/phenylpropanoids. Acta Horticulturae Sinica, 48 (9):1815-1826. (in Chinese)
doi: 10.16420/j.issn.0513-353x.2020-0549
|
|
谯正林, 胡慧贞, 鄢波, 陈龙清. 2021. 花香挥发性苯/苯丙素类化合物的生物合成及基因调控研究进展. 园艺学报, 48 (9):1815-1826.
doi: 10.16420/j.issn.0513-353x.2020-0549
|
| [22] |
Wang K, Li M Y, Hakon H. 2010. Annovar:functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research, 38 (16):e164.
|
| [23] |
Wang Mengzhao. 2016. Comparison analysis of some different genes between precocious and normal trifoliate orange[Poncirus trifoliata(L.)RAF.] based on the whole genome resequencing[M. D. Dissertation]. Wuhan:Huazhong Agricultural University. (in Chinese)
|
|
王蒙召. 2016. 基因组重测序基础上的早实枳与普通枳部分差异基因比较分析[硕士论文]. 武汉:华中农业大学.
|
| [24] |
Wang Zhiyu, Chang Beibei, Liu Qi, Cheng Xiaofan, Du Xiaoyun, Yu Xiaoli, Song Laiqing, Zhao Lingling. 2022. Study on expression and anthocyanin accumulation of solute carrier gene MdSLC35F2-like in apple. Acta Horticulturae Sinica, 49 (11):2293-2303. (in Chinese)
doi: 10.16420/j.issn.0513-353x.2021-0764
|
|
王智宇, 常贝贝, 刘琦, 程晓帆, 杜晓云, 于晓丽, 宋来庆, 赵玲玲. 2022. 苹果溶质转运蛋白基因MdSLC35F2-like表达与花青苷积累的研究. 园艺学报, 49 (11):2293-2303.
doi: 10.16420/j.issn.0513-353x.2021-0764
|
| [25] |
Wei Xiao, Zhang Qiuping, Lü Chunjing, Liu Jing, Wang Jie, Yang Wei. 2022. Genome variation detection analysis of‘Xiangjiaoli’and its late-ripening bud sport mutation based on resequencing. Molecular Plant Breeding, http://kns.cnki.net/kcms/detail/46.1068.s.20220624.1404.004.html. (in Chinese)
|
|
魏潇, 章秋平, 吕春晶, 刘晶, 王杰, 杨巍. 2022. 基于重测序的‘香蕉李’及其晚熟芽变全基因组变异分析. 分子植物育种, http://kns.cnki.net/kcms/detail/46.1068.s.20220624.1404.004.html.
|
| [26] |
Wu X M, Wu S F, Ren D M, Zhu Y P, He F C. 2007. The analysis method and progress in the study of codon bias. Hereditas, 29 (4):420-426.
|
| [27] |
Xu Q, Chen L L, Ruan X A, Chen D J, Zhu A D, Chen C L, Bertrand D, Jiao W B, Hao B H, Lyon M P, Chen J J, Gao S, Xing F, Lan H, Chang J W, Ge X H, Lei Y, Hu Q, Miao Y, Wang L, Xiao S X, Biswas M K, Zeng W F, Guo F, Cao H B, Yang X M, Xu X W, Cheng Y J, Xu J, Liu J H, Luo O J, Tang Z H, Guo W W, Kuang H H, Zhang H Y, Roose M L, Niranjan N, Deng X X, Ruan Y J. 2013. The draft genome of sweet orange(Citrus sinensis). Nature Genetics, 45 (1):59-66.
|
| [28] |
Xi F F, Guo L L, Yu Y H, Wang Y, Li Q, Zhao H L, Zhang G H, Guo D L. 2017. Comparison of reactive oxygen species metabolism during grape berry development between‘Kyoho’and its early ripening bud mutant‘Fengzao’. Plant Physiology and Biochemistry,118:634-642.
|
| [29] |
Zeng W F, Tan B, Deng L, Wang Y, Aslam M M, Wang X B, Qiao C K, Wang Z Q, Feng J C. 2020. Identification and expression analysis of abscisic acid signal transduction genes during peach fruit ripening. Scientia Horticulturae,Doi: 10.1016/j.scienta.2020.109402.
|
| [30] |
Zhang K K, Liu Z J, Guan L, Zheng T, Jiu S T, Zhu X D, Jia H F, Fang J G. 2018. Changes of anthocyanin component biosynthesis in‘Summer Black’grape berries after the red flesh mutation occurred. Journal of Agricultural and Food Chemistry, 66 (35):9209-9218.
|
| [31] |
Zhang Yin, Hu Luyan, Wang Shuming, Jing Danlong, Guo Qigao, Liang Guolu. 2023. Research advances in ABA-mediated fruit ripening. Acta Horticulturae Sinica, 50 (9):1889-1898. (in Chinese)
doi: 10.16420/j.issn.0513-353x.2023-0288
|
|
张印, 胡路艳, 王淑明, 景丹龙, 郭启高, 梁国鲁. 2023. ABA调控果实成熟研究进展. 园艺学报, 50 (9):1889-1898.
doi: 10.16420/j.issn.0513-353x.2023-0288
|
| [32] |
Zhao Yutong, Wang Wei, Mu Jiaqi, Yang Haiqing, Liu Yueping. 2022. Analysis of hormone-related gene expression patterns during fruit ripening of Prunus persica L. Journal of Beijing University of Agriculture, 37 (3):1-8. (in Chinese)
|
|
赵雨桐, 王巍, 穆佳琪, 杨海清, 刘悦萍. 2022. 桃果实成熟过程中激素相关基因表达模式分析. 北京农学院学报, 37 (3):1-8.
|